PROTEIN-SMALL MOLECULE INTERACTIONS
Docking small molecules to a protein is a fundamental step in structure-based drug design. The main approaches are (A) Docking of potential ligands from a compound database, and (B) mapping the protein for the binding sites of molecular probes - small molecules and functional groups - and using the favorable positions for the construction of larger ligands. We develop and apply algorithms for both approaches.Our basic methodology of docking is very similar to the one we have used for protein-protein docking, and consists of the following steps: (1): Rigid body search to generate a large number of conformations with good shape complementarity, and possibly favorable electrostatics and desolvation, (2) refinement, rescoring and possibly filtering using a more accurate free energy function, (3) clustering of the retained structures, and ranking the clusters on the basis of the average free energy. This algorithm has been implemented for the mapping of proteins using organic solvents as probes, and is being extended to more mainstream docking applications.
Analyzing Allosteric Sites in G Protein Coupled Receptors
The vital role of G protein-coupled receptors (GPCRs) in the homeostasis and disease biology makes this class of proteins one of the most popular classes of drug targets. Allosteric modulation of GPCRs represents a promising mechanism of pharmacological intervention. Dramatic developments witnessed in the structural biology of membrane proteins continue to reveal that the binding sites of allosteric modulators are widely distributed, including along protein surfaces. Investigating the wide range of allosteric sites identified at different locations in the 28 crystal structures of 17 receptors from four classes of GPCR targets, we showed (Wakefield et al. 2018) that both algorithms ranked the experimental allosteric site mostly within their top three sites. These predictions represent 71% and 75% of the cases for FTMap and FTSite respectively. The methods were also able to find partially hidden allosteric sites that were not fully formed in X-ray structures crystallized in the absence of allosteric ligands. These results confirm that the intrahelical sites capable of binding druglike allosteric modulators are among the strongest ligand recognition sites in a large fraction of GPCRs and suggest that both FTMap and FTSite are useful tools for identifying allosteric sites and to aid in the design of such compounds in a range of GPCR targets.
The success of FTMap inspired us to work on characterizing the conservation of allosteric sites in GPCRs. With the addition of all GPCR crystal structures to our dataset we were able to detect possible allosteric sites despite large differences in RMSDs, sequence similarities and classes. These results are both surprising and encouraging and have the potential to identify new binding sites and provide insight into the level of conservation of binding sites in GPCRs.
Many allosteric sites have been identified throughout the GPCR transmembrane region. This video shows a representative GPCR structure with 14 allosteric ligands that have been co-crystalized to Class A GPCRs.
Analysis of cryptic ligand binding sites
Molecular dynamics (MD) simulations of proteins reveal the existence of many transient surface pockets. However, it is not clear which small subset of these represents druggable or functionally relevant ligand binding sites, called “cryptic sites”. The problem is important, because cryptic allosteric sites can provide novel drug targets. We studied multiple X-ray structures for a set of proteins with validated cryptic sites using the mapping program FTMap. Results have shown that cryptic sites in ligand-free structures generally have a strong binding energy hot spot very close by. As expected, regions around cryptic sites exhibit above-average flexibility, detected as high average local RMSD between different unbound structures of the same protein. The strong hot spot neighboring each cryptic site is almost always exploited by the bound ligand, suggesting that binding to most cryptic sites involves a substantial induced fit component. We also studied the mechanisms of cryptic sites opening. It was shown that relatively few proteins have binding pockets that never form without ligand binding. Sites that are cryptic in some structures but spontaneously form in others are also rare. In most proteins the forming of pockets is impacted by mutations or ligand binding at locations far from the cryptic site. To explore these mechanisms, we applied adiabatic biased molecular dynamics (ABMD) simulations to guide the proteins from their ligand-free structures to ligand-bound conformations, and studied the distribution of druggability scores of the pockets located at the cryptic sites. Our research goal in this area is finding novel allosteric sites on potential drug target proteins. The work toward this goal will include collaboration with the Emili lab using mass spectroscopy to identify, in a high-throughput manner, metabolites that interact physically with specific cellular proteins. Our computational methods will be employed to explore the putative interactions suggested by the pull-down experiments.

Figure adapted from Z. Sun and A. Wakefield et. al (2019)
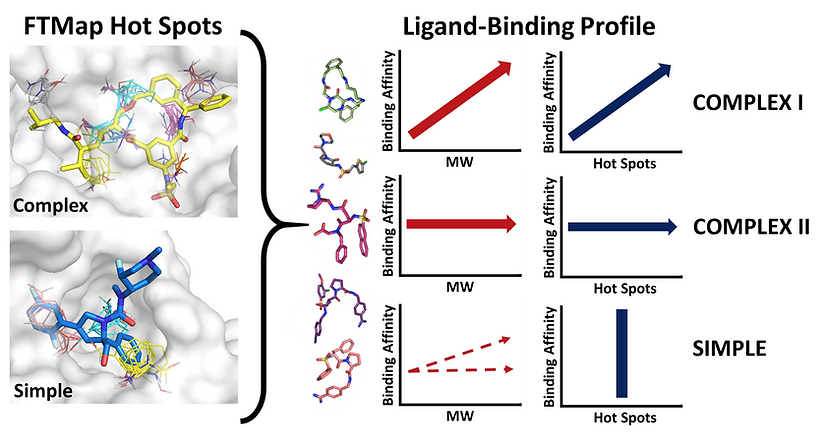
Targets that Benefit from Beyond Rule-of-5 Drugs
While an increasing number of drug candidates violate the well-known rule of five (Ro5) (Lipinski et al, 1997), it is not always clear why certain targets require or benefit from beyond Ro5 (bRo5) compounds. We performed a target- and ligand-centric analysis on the 39 protein targets identified by Doak et al, 2015 that bind a bRo5 drug or clinical candidate. First, the targets are characterized in terms of their hot spots, determined via FTMap. The hot spots represent small regions of the protein that contribute a disproportionate amount to the binding free energy. The target’s binding sites fall into one of two classifications: ‘complex’ and ‘simple’. Complex binding sites consist of multiple (n > 4), strong hot spots whereas simple binding sites have fewer (n ≤ 3) and weaker hot spots. Next, the targets are analyzed based on how different ligands interact with the protein and its hot spots, where structural data and affinity of protein-ligand complexes are available. This step reveals three different patterns for targets with complex binding sites (Complex I, Complex II, and Complex III) and one pattern for targets with simple binding sites. Complex I targets can bind both Ro5 and bRo5 ligands with high affinity, but bRo5 compounds are generally required to cover the entire hotspot complex, and the motivation for such large complexes appears to be improved binding affinity and pharmaceutical properties. Complex II targets are largely protein kinases, where the complex binding site is non-specific, and bRo5 compounds are beneficial because they provide improved selectivity. Complex III targets primarily bind only bRo5 compounds because they compensate for a number of unique binding site characteristics, such as compound induced protein stability (HIV-1 Protease) and distantly separated hot spots (Bcl-2). Finally, bRo5 compounds provide an advantage for Simple targets by extending beyond the limited binding hot spots and interacting with the walls of the protein to gain improved affinity and selectivity. Thus, the classifications we describe show the utility of bRo5 compounds in targets which have incredibly strong and defined binding sites, in addition to the traditionally considered bRo5 targets which have large, flat and featureless binding sites. The results of this study have recently been published: Egbert et al, 2019.